

Accessibility disclaimer: To obtain information contained in document files on this page in an accessible format please contact the IU Human Research Protection Program (HRPP) at (317) 274-8289 or via email at irb@iu.edu
HRPP Policy - Humanitarian use devices
About This Policy
- Effective date:
- 07/19/2018
- Last updated:
- 01/19/2021
- Policy Contact:
IU Human Research Protection Program (HRPP)
(317) 274-8289
irb@iu.edu
1.0 - Scope
This policy applies to the use of a Humanitarian Use Device (HUD) for treatment or diagnosis by a clinician at an Indiana University or IU-affiliated healthcare facility for which the IU Institutional Review Board generally provides oversight.
This policy does not apply to clinical investigational use of a HUD (see Figure 1). Use of a HUD is considered to be part of a clinical investigation if safety and effectiveness data will be collected on the HUD for the purpose of supporting a premarket approval (PMA) application. If use of a HUD is considered part of a clinical investigation, the use is considered research and research personnel must follow all policies and procedures applicable to human subjects research.
This policy does not apply to situations in which use of a HUD is necessary to save the life or protect the well-being of a patient and there is not sufficient time to request IRB approval (see Figure 1). In these situations, clinicians must follow the procedures outlined in the IU HRPP Policy on Emergency Use of Investigational Test Articles.
Back to top2.0 - Policy Statement
A HUD may only be administered in facilities under the oversight of an IRB acting in accordance with 21 CFR 56, including continuing review of the device, and only when use of the HUD has been approved by the IRB. In order to utilize a HUD for treatment or diagnosis, IRB approval must be obtained prior to use. Once approved, the HUD may be used only for the indications approved by the IRB.
In reviewing a request for HUD use, the IRB must consider the risks to patients as described in the product labeling, ensure the risks are minimized, and evaluate whether the risks are reasonable in relation to the proposed use of the device. The IRB has the discretion to approve use of the HUD for off-label indications given sufficient justification from the clinician(s).
Figure 1 – Overview of regulatory procedures Reference: FDA Guidance: Humanitarian Device Exemption (HDE) Regulation: Questions and Answers; Guidance for HDE Holders, Institutional Review Boards (IRBs), Clinical Investigators, and Food and Drug Administration Staff, issued July 8, 2010 (Guidance retired but figure is still applicable)
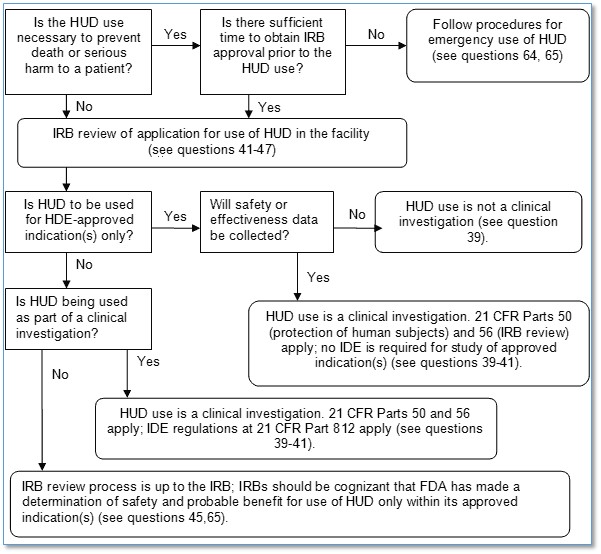
Description of Figure 1 - Overview of regulatory procedures
If HUD use is necessary to prevent death or serious harm to a patient
- If there is time to obtain IRB approval prior to the HUD use, submit for IRB review for use of the HUD in the facility
- If there is not time to obtain IRB approval, follow procedures for emergency use of HUD
If HUD use is not necessary to prevent death or serious harm to a patient:
- Submit for IRB review for use of the HUD in the facility
If HUD will be used for HDE-approved indications only:
- If safety or effectiveness data will not be collected, HUD use is not a clinical investigation
- If safety or effectiveness data will be collected, HUD use is a clinical investigation; 21 CFR Parts 50 and 56 apply; no IDE is required for study of approved indications
If HUD use is not limited to HDE-approved indications only:
- If HUD is used as part of a clinical investigation, HUD use is a clinical investigation. 21 CFR Parts 50 and 56 apply; IDE regulations at 21 CFR Part 812 apply
- If HUD is not used as part of a clinical investigation, IRB review process is up to the IRB; IRBs should be cognizant that FDA has made a determination of safety and probable benefit for use of HUD only within its approved indications
The use of a HUD must be initially reviewed at a convened IRB meeting and approved before the device can be used. To facilitate initial review, the HUD application must be submitted via Kuali Protocols, with the following documentation attached:
- The HUD manufacturer's product labeling, clinical brochure, and/or other pertinent manufacturer informational materials
- HUD manufacturer's patient information packet, if available, which should be provided to patients
- The FDA HDE approval letter
Research informed consent and authorization documents are not applicable and should not be submitted, as use of a HUD under this policy is not considered research.
All facilities which will utilize the HUD and any clinician wishing to use the device must be included in the application in Kuali Protocols.
The IRB must conduct continuing review/renewal of the use of the HUD within the appropriate time frame as specified by the IRB at time of initial review, or use of the HUD must cease until such time that it can be reviewed. Renewal information must be submitted via Kuali Protocols and review may be conducted via the expedited procedure.
Clinicians should track and report the following at the time of renewal:
- The number of patients who received the HUD since the beginning of the study
- Summary of minor deviations, minor noncompliance, and/or noncompliance that required prompt reporting but was determined by HRPP staff to not be apparent serious or continuing noncompliance since the last IRB review
- Statement whether adverse events occurred in excess of the expected frequency and level of severity as documented in the HUD manufacturer's product labeling, clinical brochure, and/or other pertinent manufacturer informational materials and, if so, confirmation that the event was promptly reported
- Summary of any patient complaints
- Any new information that may be relevant in assessing impact on patient safety or continued use of the HUD, including literature publications, audit/monitoring findings, and/or interim findings.
If modifications to the initial IRB approval are needed to allow for additional indications, either FDA-approved or justified by the clinicians, the clinician(s) should submit an amendment to the existing protocol via Kuali Protocols describing the modifications and the rationale.
Reportable Events must be reported to and reviewed by the IRB in accordance with the IU HRPP Policy on Reportable Events.
4.0 - Sanctions
Individuals found to be in violation of this policy may be subject to sanctions related to the clinical use of the HUD at Indiana University's affiliates, including withdrawal of IRB approval.
Back to top5.0 - History
Policy contact updated from HSO to HRPP. Scope statement revised to reflect this Policy applies to clinicians at an Indiana University or IU-affiliated healthcare facility. KC IRB replaced with Kuali Protocols throughout. Section 3.2 revised to reflect information clinician should track and report at time of renewal. Removed reference to KC IRB and replaced FDA Guidance with newer version in section 6.0. Relevant definitions added to section 7.0.
Back to topIU HRPP Policies
Related Policy Documents
View Expanded Access of Investigational Test Articles for Single-Patient Use Policy
IU HRPP Guidance
Related Guidance Documents
N/A
Regulatory References
7.0 - Definitions related to this policy
adverse events, audit, clinical investigation, device, humanitarian device exemption (HDE), humanitarian use device, institutional review board (IRB), investigator, noncompliance, reportable event
View All Abbreviations and Definitions
Back to top